.jpg)
Interleukin-6 und rheumatoide Arthritis
IL-6 spielt eine Schlüsselrolle in der Pathophysiologie der RA.
Die rheumatoide Arthritis (RA) ist eine chronische Systemerkrankung, die durch ein komplexes Netzwerk von Zytokinen ausgelöst und vorangetrieben wird. Interleukin-6 (IL-6) ist eines der Zytokine, die am häufigsten im Serum und in der Synovialflüssigkeit der entzündeten Gelenke von RA-Patienten vorkommen. Die Spiegel an IL-6 und löslichem IL-6-Rezeptor in der Synovialflüssigkeit korrelieren signifikant mit der Schwere der Gelenkzerstörung.¹
Unter normalen physiologischen Umständen übt IL-6 u. a. lebenswichtige proinflammatorische Funktionen als Reaktion auf Infektionen oder Verletzungen aus.¹⁻³ Eine dauerhaft erhöhte IL-6-Konzentration hingegen kann pathologische Auswirkungen haben und gehört zu den Hauptauslösern für das dysfunktionale und chronisch entzündliche Milieu bei RA.³⁻⁵
Welche artikulären RA-Manifestationen werden durch IL-6 beeinflusst?
IL-6 spielt eine zentrale Rolle bei den artikulären Manifestationen der RA.¹,⁶–⁸ Hier ist IL-6 zentral an der Mediation der chronischen Entzündung beteiligt und induziert unter anderem die Gelenk- und Knochenzerstörung über Pannusbildung und Osteoklastenaktivierung.¹
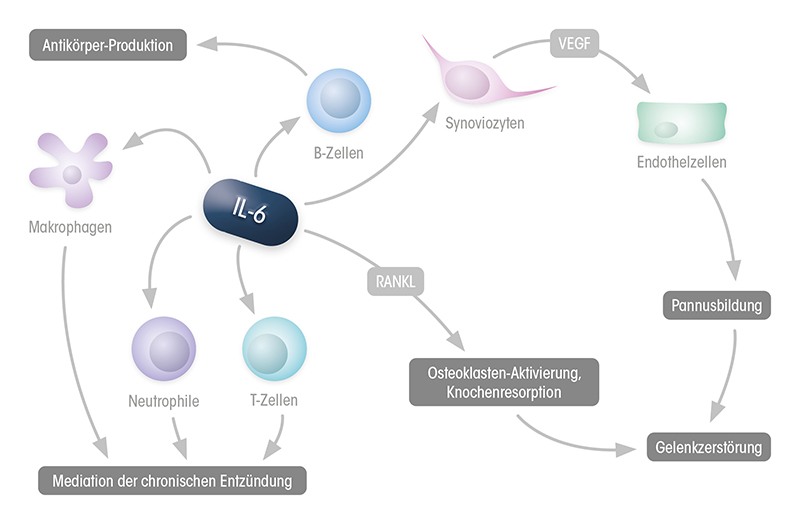
Zusammenspiel von infiltrierenden Immunzellen, Synoviozyten und Endothelzellen im entzündeten Gelenk,
VEGF: Vascular Endothelial Growth Factor; RANKL: Receptor Activator of NF-κB Ligand (modifiziert nach 1)
- Unter normalen Umständen sind die Spiegel von IL-6 gering. Basierend auf mehreren Untersuchungen zeigten sich Serumspiegel von 1–16 pg/ml IL-6 im Blutkreislauf gesunder Personen.²⁹–³⁴
- Patienten mit RA jedoch haben Serum-IL-6-Spiegel von 5–200 pg/ml mit 100–1000-fach höheren Konzentrationen in der Synovialflüssigkeit.⁷,³⁰,³¹,³³,³⁵–³⁷
- In Übereinstimmung damit ist IL-6 eines der am häufigsten vorkommenden Zytokine in Serum und Synovialflüssigkeit von RA-Patienten und korreliert sowohl mit Krankheitsaktivität als auch Gelenkzerstörung.¹,⁷,³⁸–⁴⁰
- Insbesondere am frühen Morgen zeigten sich bei RA-Patienten bezüglich der IL-6 Serumkonzentrationen Spitzenwerte im Vergleich zu gesunden Kontrollpersonen.⁴¹
- Proinflammatorische Zellen und Mediatoren wie z. B. neutrophile Granulozyten, Makrophagen, Fibroblasten-ähnliche Synoviozyten (FLS), T- und B-Zellen werden durch IL-6 in die Gelenke rekrutiert bzw. innerhalb der Gelenke aktiviert.⁸–⁹,¹⁷
- Dies führt zu einer verstärkten Entzündung und letztendlich zu strukturellen Gelenkschäden.21
- Der Knorpel wird durch die Aktivierung von FLS und Chondrozyten, die Cathepsine und Matrix-Metalloproteinasen exprimieren, abgebaut.¹⁸,²¹–²⁴
- IL-6 aktiviert und erhöht die Proliferation der FLS in der Synovialmembran.
- Osteoklastengenese und Osteoklastenaktivität werden stimuliert und führen zu strukturellen Schädigungen durch Knochenresorption. Darüber hinaus existieren Hinweise, dass IL-6 und/oder der lösliche IL-6-Rezeptor (sIL-6-R) an der Regulierung von Osteoklasten-Vorläuferzellen im Knochenmark (hämatopoetische Stammzellen) vor und während der entzündlichen Arthritis beteiligt ist/sind.⁷,⁸,¹⁴,¹⁸–¹⁹
Die IL-6-Serumkonzentrationen sind am frühen Morgen am höchsten – zu dieser Zeit treten bei RA-Patienten am häufigsten Gelenkschmerzen und Gelenksteifigkeit (Morgensteifigkeit) sowie Funktionsbeeinträchtigungen auf.
Dauerhaft erhöhte IL-6-Konzentrationen halten eine chronische Synovitis aufrecht:*,¹,⁹,¹⁰
* basierend auf präklinischen und klinischen Daten und Ex-vivo-Daten.
IL-6-aktivierte Fibroblasten-ähnliche Synoviozyten (FLS) spielen eine Schüsselrolle bei der chronischen Entzündung und der Gelenkzerstörung bei RA.²⁴–²⁷
Welche systemischen RA-Manifestationen werden durch IL-6 beeinflusst?
Neben der zentralen Rolle im entzündeten Gelenk ist IL-6 auf der systemischen Ebene unter anderem an der Akute-Phase-Reaktion, der Steuerung des Eisen- und Lipid-Stoffwechsels sowie der Regulierung der HPA-Achse (Hypothalamus-Hypophysen-Nebennieren-Achse) beteiligt.¹,²⁵,⁴² Aufgrund dessen können erhöhte IL-6 Konzentrationen nicht nur zu artikulären Manifestationen, sondern auch zu vielfältigen systemischen Symptomen führen.
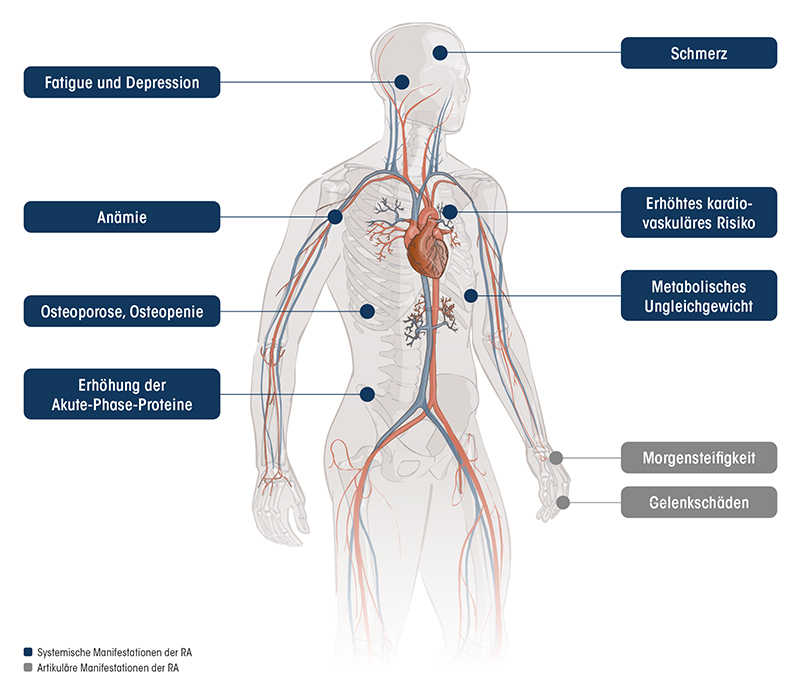
Durch IL-6 vermittelte artikuläre und systemische RA-Manifestationen¹,²⁵,⁴²
- Immunzellen
- Synoviale Fibroblasten
- Hämatopoetische Stammzellen
- Hepatozyten
- Adipozyten
- Endothelzellen
- Langerhans-Zellen
- Fatigue²⁵
- Depressionen, depressive Verstimmungen²⁵
- Beeinflussung des Eisenstoffwechsels durch Induktion von Hepcidin²⁵
- Erhöhte Hepcidin-Spiegel sind – gerade bei chronisch-entzündlichen Erkrankungen –eine potenzielle Ursache für Anämie²⁵
- Osteoklastenaktivierung²⁵
- Allgemeiner Knochendichteverlust¹⁴
- Systemische Entzündung durch Wirkung auf die Leber, die zu einer Erhöhung von C-reaktivem Protein (CRP) und Serum-Amyloid-A (SAA) führt²⁵
- CRP-Erhöhung – was mit einem erhöhten Risiko für kardiovaskuläre Erkrankungen assoziiert ist¹
- Beeinflussung der vaskulären endothelialen Dysfunktion¹,⁵¹
- Interaktionen mit dem Fettgewebe⁴⁷,⁴⁸
- Einfluss auf den LDL (low density lipoprotein)-Metabolimsus⁵⁰
- Autoantikörperbildung¹
- Dysregulation der T- und B-Zellen¹,¹⁴,¹⁶
Eine erhöhte IL-6-Signaltransduktion kann die Homöostase zahlreicher physiologischer Prozesse stören.⁶,⁴³,⁴⁴
IL-6 besitzt durch seine Interaktion mit membrangebundenen und löslichen Rezeptoren eine hohe biologische Aktivität und kann mit einer Vielzahl an Zell- und Gewebstypen interagieren.³⁻⁵,⁴¹,⁴⁴⁻⁴⁸
Dazu gehören insbesondere:
IL-6 trägt zur Zerstörung der Gelenke bei und wird mit den systemischen Manifestationen der RA in Verbindung gebracht.²⁵,⁴¹,⁴⁹
Erhöhte IL-6-Konzentrationen wirken auf verschiedene physiologische Prozesse und können dadurch u. a. zu Müdigkeit, Anämie, Osteoporose und Herz-Kreislauf-Erkrankungen führen.²⁵,⁴¹,⁴⁶
Einfluss auf die Psyche:
Anämie:
Osteoporose, Osteopenie:
Erhöhung der Akute-Phase-Proteine:
Erhöhtes kardiovaskuläres Risiko:
Metabolisches Ungleichgewicht:
Einfluss auf das Immunsystem:
Literatur:
1. Dayer JM, Choy E Rheumatology (Oxford) 2010; 49(1): 15–24.
2. McInnes IB. Cytokines. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR, eds. Kelley’s Textbook of Rheumatology. Vol 1. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013: 369–81.
3. Saxena A, Cronstein BN. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR, eds. Kelley’s Textbook of Rheumatology. Vol 1. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013: 818–29.
4. Tutuncu Z, Kavanaugh A. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O’Dell JR, eds. Kelley’s Textbook of Rheumatology. Vol 1. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013: 957–77.
5. Tanaka T, Kishimoto T. Int J Biol Sci 2012; 8(9): 1227–36.
6. Ito A et al. Arthritis Rheum 1992; 35(10): 1197–1201.
7. Kotake S et al. J Bone Miner Res 1996;11(1): 88–95.
8. Wong PK et al. Arthritis Rheum 2006; 54(1): 158–68.
9. Choy E. Curr Rheumatol Rep 2008; 10(5): 413–7.
10. Sack U et al. Rheumatol Intl 1993; 13(2): 45–51.
11. Nakamura I et al. J Rheumatol 2009; 36(2): 459–60.
12. van Leeuwen MA et al. Ann Rheum Dis 1995; 54(1): 33–8.
13. Hashizume M et al. Rheumatology (Oxford) 2008; 47(11): 1635–40.
14. Maggio M et al. J Gerontol A Biol Sci Med Sci 2006; 61(6): 575–84.
15. Chomarat P et al. Nat Immunol 2000;1(6): 510–4.
16. de Hooge AS et al. Am J Pathol 2000; 157(6): 2081–91.
17. Ducreux J et al. Arthritis Rheumatol 2014; 66(1): 15–23.
18. Garnero P, Thompson E et al. Arthritis Rheum 2010; 62(1): 33–43.
19. Ishimi Y et al. J Immunol 1990; 145(10): 3297–303.
20. Palmqvist P et al. J Immunol 2002; 169(6): 3353–62.
21. Choy E et al. Nat Rev Rheumatol 2013; 9(3): 154–63.
22. Firestein GS Arthritis Rheum 1996; 39(11): 1781–90.
23. Rowan AD et al. Arthritis Rheum 2001; 44(7): 1620–32.
24. Bartok B, Firestein GS Immunol Rev 2010; 233(1): 233–55.
25. Choy E. Rheumatology (Oxford) 2012; 51(suppl 5): v3–v11.
26. McInnes IB, Schett G. Nat Rev Immunol 2007;7(6): 429–42.
27. Bottini N, Firestein GS. Nat Rev Rheumatol 2013; 9(1): 24–33.
28. Matsumoto T et al. Rheumatol Int 2006; 26(12): 1096–100.
29. Fischer CP. Exerc Immunol Rev 2006; 12: 6–33.
30. Desgeorges A et al. J Rheumatol 1997; 24: 1510–6.
31. Sack U et al. Rheumatol Int 1993; 13: 45–51.
32. Pujhari SK et al. J Neuroimmunol 2013; 263: 133-8.
33. Uson J et al. J Rheumatol 1997; 24: 2069–75.
34. Motivala SJ et al. Psychosom Med 2005; 67: 187–94.
35. Hein GE et al. Rheumatol Int 2005; 26: 137–41.
36. Sacerdote P et al. Inflamm Res 1995; 44: 486–90.
37. Abe H et al. Rheumatology (Oxford) 2014; 53: 165–72.
38. Firestein GS et al. J Immunol 1990; 144(9): 3347–53.
39. Madhok R et al. Ann Rheum Dis 1993; 52(3): 232–4.
40. Baillet A et al. Arthritis Care Res (Hoboken) 2015; 67(7): 905–12.
41. Crofford LJ et al. J Clin Endocrinol Metab 1997; 82(4): 1279-83.
42. Srirangan S, Choy EH. The role of interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther Adv Musculoskel Dis 2010; 2(5): 247–56.
43. Rose-John S et al. J Leukoc Biol 2006; 80(2): 227–36.
44. Bode JG et al. Eur J Cell Biol 2012; 91(6-7): 496–505.
45. Okada A et al. Clin Exp Rheumatol 2012; 30(3): 332–37.
46. Colmegna I et al. Clin Pharmacol Ther 2012; 91(4): 607–20.
47. Trujillo ME et al. J Clin Endocrinol Metab 2004; 89(11): 5577–82.
48. Hashizume M, Mihara M. Arthritis 2011; 765624
49. Hochberg MC et al. eds. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier Ltd; 2011.
50. Gierens H et al. Arterioscler Thromb Vasc Biol 2000; 20(7): 1777–83.
51. Sattar N et al. Circulation 2003; 108(24): 2957–63
MAT-DE-2000485-2.0-06/2023